OpenPandemics: COVID-19 Update (July 2020)
 In the past months, the team worked very hard to get our OpenPandemics: COVID-19 project started, in collaboration with the IBM World Community Grid.
In the past months, the team worked very hard to get our OpenPandemics: COVID-19 project started, in collaboration with the IBM World Community Grid.
The goal of the project is to virtually screen very large collections of chemical compounds to identify potential inhibitors of the SARS-CoV2 virus that can provide a starting point for the development of new therapeutic tools for COVID-19. After facing a number of challenges, we finally launched the project and we already collected many results that are currently analyzed. Every single compound that we will identify in our virtual screenings will be experimentally tested in in vitro assays by our collaborators (at Scripps and elsewhere), and hopefully evolve to become COVID-19 antivirals. Our project is one of the several active efforts that are trying to find new potential drugs for fighting this pandemic. What is unique about our project is that we are using new molecular docking protocols, together with more conventional approaches, that allow us to target specific residues and regions of the viral proteins in a way that is very difficult for the virus to escape.
Until now, we have been very busy setting up the software infrastructure, preparing the input data for the volunteers to process, and get ready to receive and analyze the huge amount of data that is going to be returned.
Thank you all for your contribution!
This is a (long overdue!) summary for all the volunteers, covering the virtual screening strategy and details about the SARS-CoV-2 targets that have been considered so far for the OpenPandemics: COVID-19 project. This is an ongoing work and more structures will be targeted and we will keep you updated on the new included structures and on the first results coming in as well.
Ligand libraries (Diogo, Matthew)
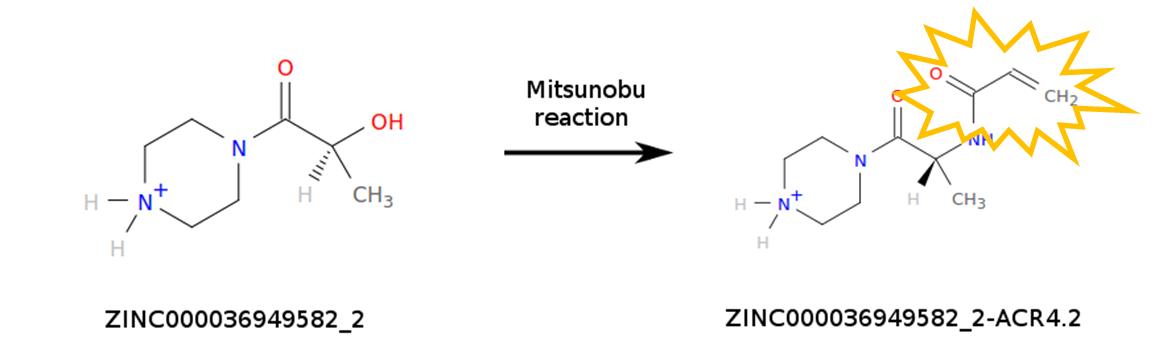 The computations carried out by World Community Grid (WCG) volunteers simulate millions of chemical compounds to help identify those that may interact with SARS-CoV-2 proteins. While there are several commercial sources for compounds, we decided to simulate not only molecules that are available for purchase, but also molecules that can be made by modifying existing ones. In particular, we considered chemical reactions that insert a chemical warhead that is reactive enough to react with the viral proteins, but not too much to become dangerously toxic. The purpose of the reactive warhead is to form a covalent bond with the viral proteins, making the binding event irreversible. We considered only simple chemical reactions that can be performed by our collaborators in a short amount of time. At the moment, WCG volunteers are simulating a library of 37 million compounds containing the acrylamide warhead which is specific to cysteine residues. New compounds will be added to the library in the upcoming months.
The computations carried out by World Community Grid (WCG) volunteers simulate millions of chemical compounds to help identify those that may interact with SARS-CoV-2 proteins. While there are several commercial sources for compounds, we decided to simulate not only molecules that are available for purchase, but also molecules that can be made by modifying existing ones. In particular, we considered chemical reactions that insert a chemical warhead that is reactive enough to react with the viral proteins, but not too much to become dangerously toxic. The purpose of the reactive warhead is to form a covalent bond with the viral proteins, making the binding event irreversible. We considered only simple chemical reactions that can be performed by our collaborators in a short amount of time. At the moment, WCG volunteers are simulating a library of 37 million compounds containing the acrylamide warhead which is specific to cysteine residues. New compounds will be added to the library in the upcoming months.
Target structures (Jerome, Giulia, Batuu, Paolo, Martina, Christina)
In the initial phase of the project, we are focusing our effort against 3 protein targets of SARS-CoV-2: the papain-like proteases (PLpro, or nsp3), the main protease (Mpro, or nsp5) and the endoribonuclease (nsp15). After the analysis of the druggable pockets on each structure, the most promising cysteine residues, as well as catalytic cysteines, have been selected as potential covalent sites.
1. The first target we are considering is the papain-like protease (PLpro, or nsp3), which is part of the multi-domain protein nsp3. It is responsible for viral polyprotein maturation and involved in repressing the host innate immune response. In this protein, we are targeting a specific residue, cysteine 111, located in the active site and which plays a key role in the catalytic activity. Right now, we are considering two different forms of the protein, one from SARS-CoV2 and the other one from SARS-CoV1 (Protein Data Bank ids: 6w9c and 4mm3). Why so, you may ask? Well, at the time we started this project, in March, few structures were available of SARS-CoV2, and only one (PDB id: 6w9c) with the key feature we were looking for: a model of the cysteine 111 residue accessible enough to be targeted by covalent binders. Fortunately for us, the active site does not differ much between the two SARS-CoV1 and CoV2 viruses, allowing us to also exploit another structure (PDB id: 4mm3) with the right conformation more suitable for virtual screenings. And more is definitely better! Since then, many other structures of nsp3 from SARS-CoV2 were solved with other very interesting structural variations in the active site. And of course, those conformations will be incorporated in the future screenings!
This represents also an interesting challenge because we can test a key hypothesis: can we identify broad spectrum viral inhibitors that work against multiple viral strains? Knowing this is essential to estimate on how difficult it will be to tackle future pandemics.
For more in-depth details about PLpro, see the the blog post from our scientific collaborators in Germany, who provide a great service to the community by improving the quality of structures.
All the calculations that are running right now are targeting the different conformations of PLpro.
2. The Main Protease (MPro, or nsp5), which is the main target in many studies from other laboratories, is responsible for maturation of the viral polyprotein during the infection. Currently, we are considering 5 structures to take into account different conformations of the protein (PDB ids: 6y84, 5re9, 5ren, 6lu7 and 6w63). For sure, they all look the same from far away, but when you look more closely, some small structural variations can make a huge difference! For this protein, 3 cysteines were chosen as covalent binding sites: cysteine 145 located in the active site, cysteine 156 and cysteine 300 located in alternative binding sites, which have not yet been explored. For the main cysteine 145, all 5 conformations are used, but only one for cysteine 156 and cysteine 300 (PDB id: 6y84). At the end, we decided to dedicate more computational time for the active cysteine for which we know for sure it can react with covalent binders.
This will be the next target we will focus our efforts on.
3. The Nidoviral RNA uridylate-specific endoribonuclease (NendoU, or nsp15) is a conserved endoribonuclease across multiple families of RNA viruses, including coronaviruses. While initially suspected to directly participate in RNA replication, its exact roles in the case of SARS-CoV and SARS-CoV-2 are still debated even if more recently, it has been suggested to interfere with the host’s immune response against double-stranded RNA intermediates, which are generated during the replication process. After some investigations of the crystal structure of NendoU (PDB id: 6vww), residues cysteine 293 (located in the active site) and cysteine 103 have been chosen as potential covalent sites for acrylamide warheads. After the first two targets are completed, we will tackle nsp15.
Concerning the external spike glycoprotein, which is the protein responsible for fusion and entry into human host cells, we know that many of you asked why we are not currently targeting this protein. The main challenge of this protein resides in its intrinsic high flexibility. We know that the spike protein can exist in mainly two different states: open or close. So why don’t we just take these two structures? Well, things are more complicated because beside being very flexible, this protein is also covered in a complex sugar network that protects it from the host immune system, and makes it hard to target directly with small molecules. Our collaborators are working on setting up their biological assays, so we are definitely interested in providing them with molecules to test. In the meantime more structural data will be available for us to analyze and assess where more “druggable” sites can be.
Docking method: Reactive docking (Giulia)
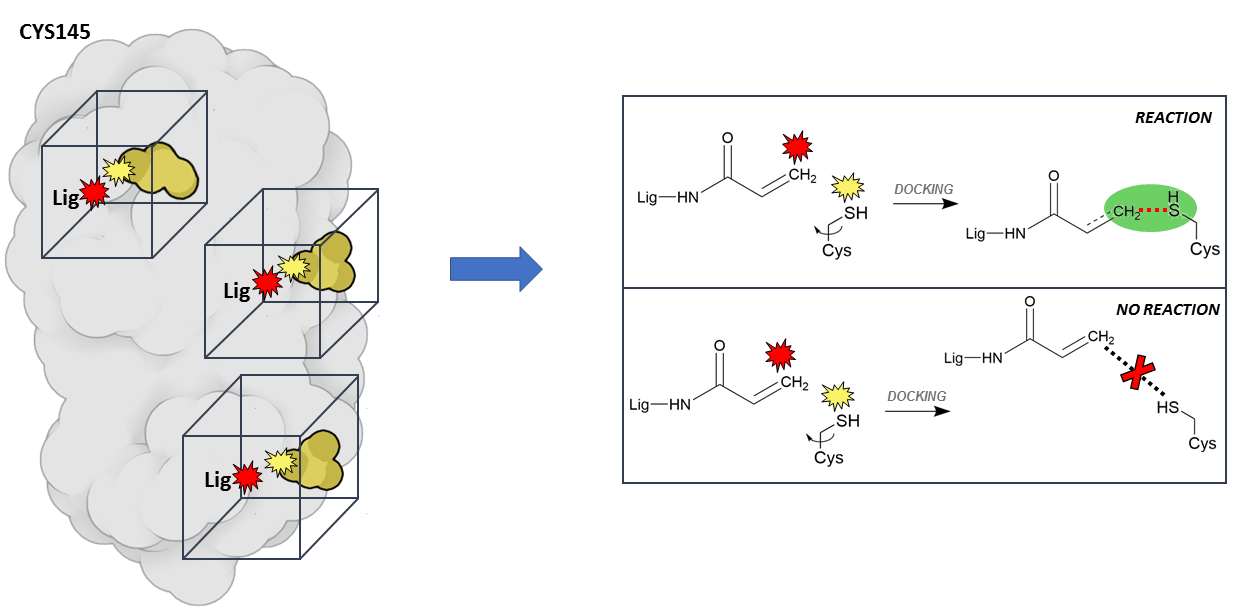 The main innovation of our approach resides in the docking protocols we are using. Together with conventional dockings, in which we try to predict which molecules can bind transiently (i.e., reversibly) on the protein surface, we are using a new protocol, called ‘reactive docking’, to find irreversible binders: molecules that can bind tighter and irreversibly. One of the advantages of this class of molecules is that once they latch on their target, they disable it until the protein is degraded, which is clearly a great feature to have in a drug! and if properly designed they can be very selective. For example, penicillin, probably the most widely known antibiotic, works exactly in this way, by reacting specifically with the wall proteins of bacteria without interacting with human proteins. The ‘reactive docking’ is a protocol we developed in our lab that allows us to simulate the reaction event between the ligand and a specific covalent residue on the protein. Essentially, every result in which the “reactive” atoms on the ligand and the residue get within bond-distance, we assume that the reaction is likely to happen. From a technical point of view, this represents an important advantage because we expect it to reduce the false positive rates (i.e., molecules wrongly predicted to be active) dramatically, since we can filter out all those that did not react. To date, this is the only docking method that allows to perform such virtual screening experiments on very large libraries of compounds.
The main innovation of our approach resides in the docking protocols we are using. Together with conventional dockings, in which we try to predict which molecules can bind transiently (i.e., reversibly) on the protein surface, we are using a new protocol, called ‘reactive docking’, to find irreversible binders: molecules that can bind tighter and irreversibly. One of the advantages of this class of molecules is that once they latch on their target, they disable it until the protein is degraded, which is clearly a great feature to have in a drug! and if properly designed they can be very selective. For example, penicillin, probably the most widely known antibiotic, works exactly in this way, by reacting specifically with the wall proteins of bacteria without interacting with human proteins. The ‘reactive docking’ is a protocol we developed in our lab that allows us to simulate the reaction event between the ligand and a specific covalent residue on the protein. Essentially, every result in which the “reactive” atoms on the ligand and the residue get within bond-distance, we assume that the reaction is likely to happen. From a technical point of view, this represents an important advantage because we expect it to reduce the false positive rates (i.e., molecules wrongly predicted to be active) dramatically, since we can filter out all those that did not react. To date, this is the only docking method that allows to perform such virtual screening experiments on very large libraries of compounds.
Data Architecture (Andreas)
Before the first results started coming in, we got ready to be able to receive all the results, store them, and make them available for filtering and analysis. Each of your runs creates about 300 KB of data in two results files – one with the DLG extension and the other a plain XML file for easier indexing. The DLG file extension is our program’s (AutoDock4.2) main result file storing the coordinates and energies of the docked ligand candidates (Docking LoG).
While 300 KB per run on average may not sound like a lot, when you multiply it by the number of runs WCG sends out per input package (about 10,000 runs) it would actually come out to about 3 GB. We started preparing more than 10,000 packages, and hope to go way beyond that eventually. With compression (good old Gzip!) we are able to get a single package down to about 400 MB. Based on early estimates about the number of runs we would end up with multiple tens of TB of data thus created.
In order to reduce the disk usage for storing such a large amount of data, we designed a method to get a single package down to about 85 MB, or about 20% of its compressed size, by utilizing the internal description of docking solutions used in our software. Typically, in dockings there is a fixed target (the so called receptor, i.e. a given protein of COVID-19) and a flexible molecule to be docked (the so called ligand, i.e. a potential drug). The ligand is moved around, rotated, and its geometry is determined by bond rotations between its atoms. We call the set of parameters that describes the aforementioned properties “genome” (since we use a genetic algorithm to run our searches… that’s for another post…), and like in actual life, a ligand genome can be used to fully describe it. By storing only the ligand “genetic information” we were able to bring the data size down to about 20% of its originally compressed size. These “dried” genomes reduce dramatically the amount of data that needs to be transferred and stored in our database.
While it takes you up to 2.8 CPU years to find the optimal genome result for a single package, it takes us only about 30 minutes to re-generate the original coordinates and energies (since we already know the solution). We have two dedicated servers for receiving and storing the data, and perform the processing operations.
So what happens to the data during the processing? There are two basic things that happen as soon as we receive a result file: first, the “dried” genomes in the results are “rehydrated”, meaning the coordinates of all poses for each run are calculated in the tridimensional space; then each pose is analyzed individually.
An analysis script collects the information about the type of interactions (e.g., polar/non- interactions, electrostatic, etc.) and the target residues with which each pose is interacting (i.e. cysteine 111). It also checks if the molecule reacted or not (according to the reactive docking parameters). This information is gathered in a very efficient way, and stored in our database. This is an essential step in the processing phase because it enables us to perform very complex queries that ultimately allow us to look for the few hundred promising “needles” in a haystack that contains billions of poses in a few seconds. The most promising compounds will be visually inspected one by one and selected to be advanced to the testing phase. Thanks to all of your support, at the time of writing the database contains about 3,600 results packages corresponding to a total of about 36 million ligands or 1.8 billion ligand poses, with more results coming in daily.
Check the latest statistics on the OpenPandemics: COVID-19 calculations.